

近年来,光氧化还原催化在太阳能转化、CO₂还原及有机合成等可持续化学领域中发挥着重要作用。铱(III)配合物因其优异的光物理特性与电子传递能力,成为光催化体系中最具代表性的催化剂之一,氧化还原电位是决定其催化活性和电子转移效率的关键参数。然而,传统实验测定(如循环伏安法)耗时、材料需求大,难以快速筛选大规模候选催化剂;量子化学计算方法(如DFT计算)虽可预测电位,但计算成本高且存在体系误差,尤其是激发态氧化还原电位预测难度大,因其涉及复杂的电子结构与短寿命态,同时缺乏统一、系统的数据集,也缺少能够平衡准确性、可解释性与高通量的预测策略。这些问题都使激发态氧化还原电位预测方面面临巨大挑战。
为解决上述问题,东北师范大学化学学院功能材料化学研究所关威教授和朱博副教授团队建立了一种结合高精度DFT计算与机器学习的可解释性预测框架,实现了对铱(III)光催化剂基态与激发态氧化还原电位的高效、可扩展预测。
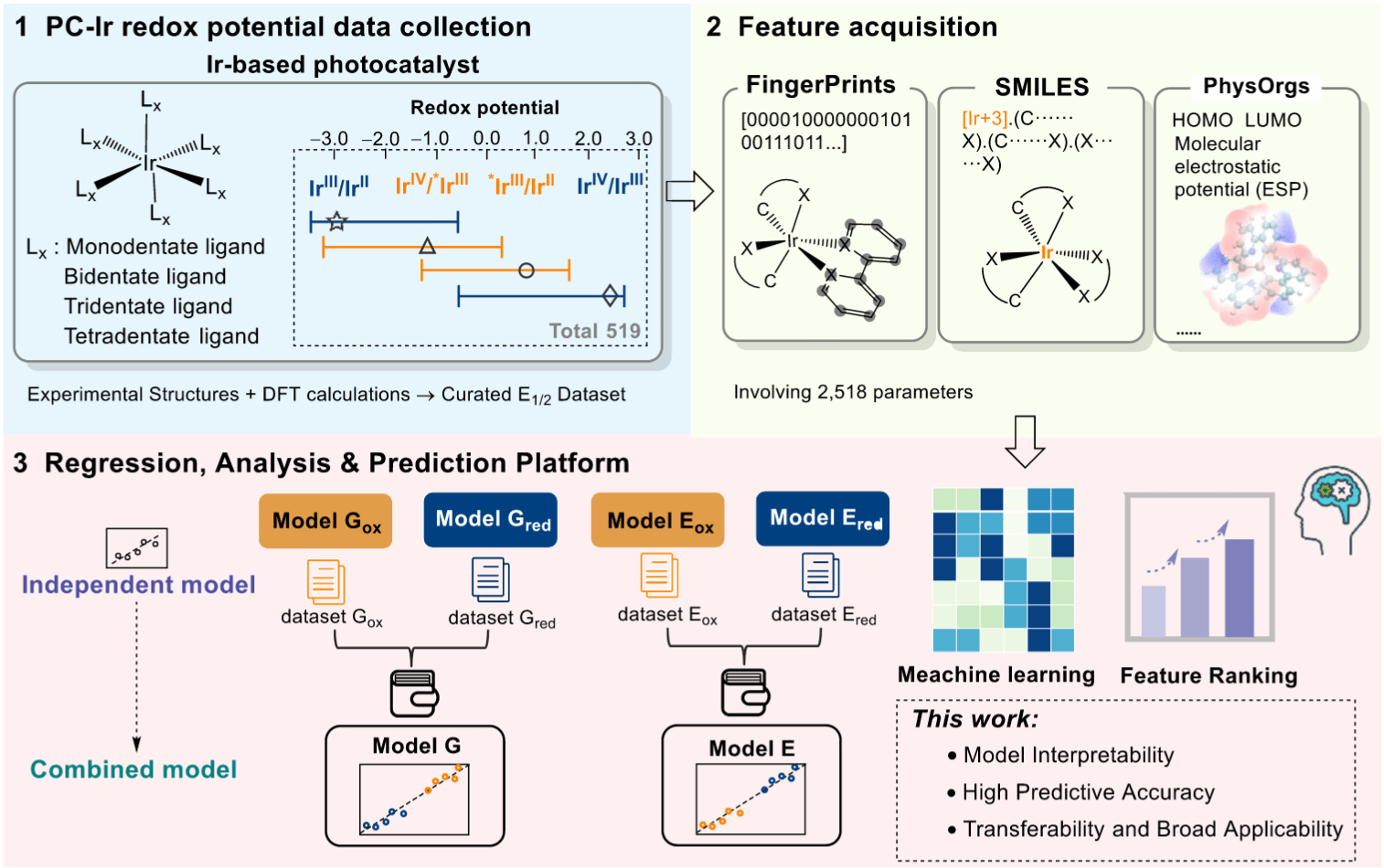
研究总体流程图展示了整个研究的计算与建模流程,概括了从数据计算到机器学习预测的完整自动化流程。模块1数据生成:通过统一的DFT计算获得铱(III)光催化剂的基态与激发态氧化/还原电位数据;模块2特征提取:提取分子的几何与电子结构描述符。模块3机器学习建模:利用回归与特征分析模型进行训练、验证和预测,实现高精度氧化还原电位预测。
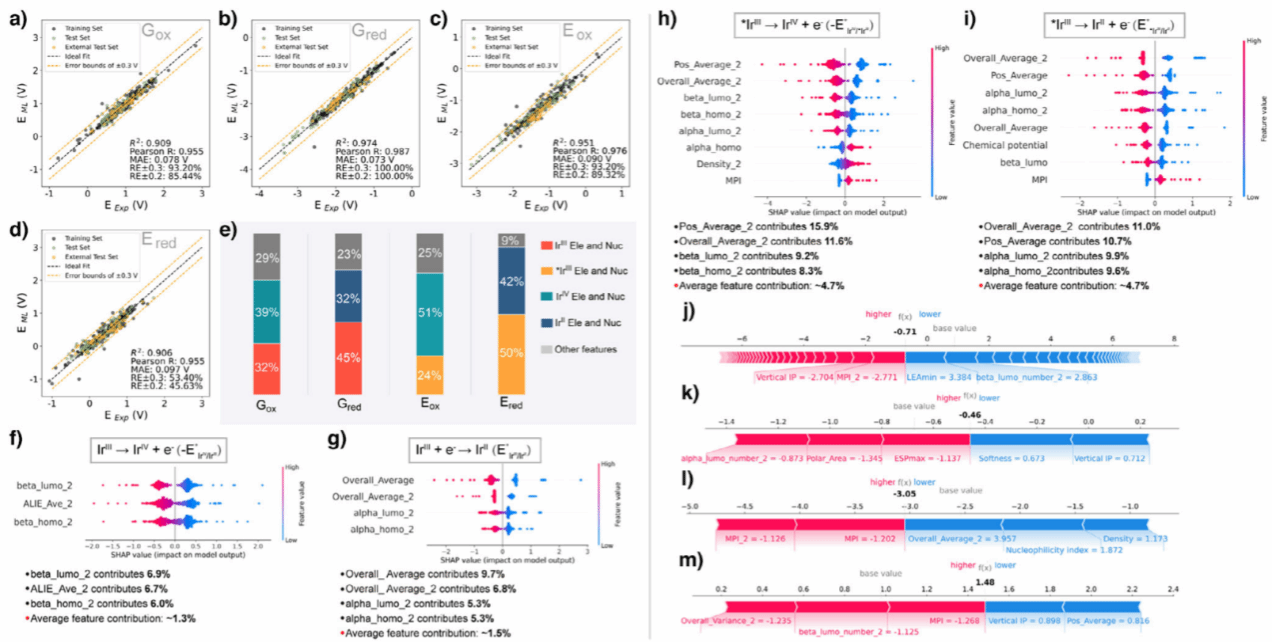
独立模型的性能与特征分析图中,展示四个独立模型(Gox、Gred、Eox、Ered)的预测结果和关键特征贡献,揭示了模型的高准确性,并用可解释分析明确了电子结构如何影响氧化还原性能。(a–d):模型预测值与参考值对比,误差大多在 ±0.3 V 以内,说明模型可靠。(e):不同模型中特征的重要性分布。(f–i):SHAP分析图,显示哪些电子特征(如HOMO/LUMO能级、电势分布)对电位影响最大。(j–m):SHAP力图示例,解释单个催化剂的具体预测原因。
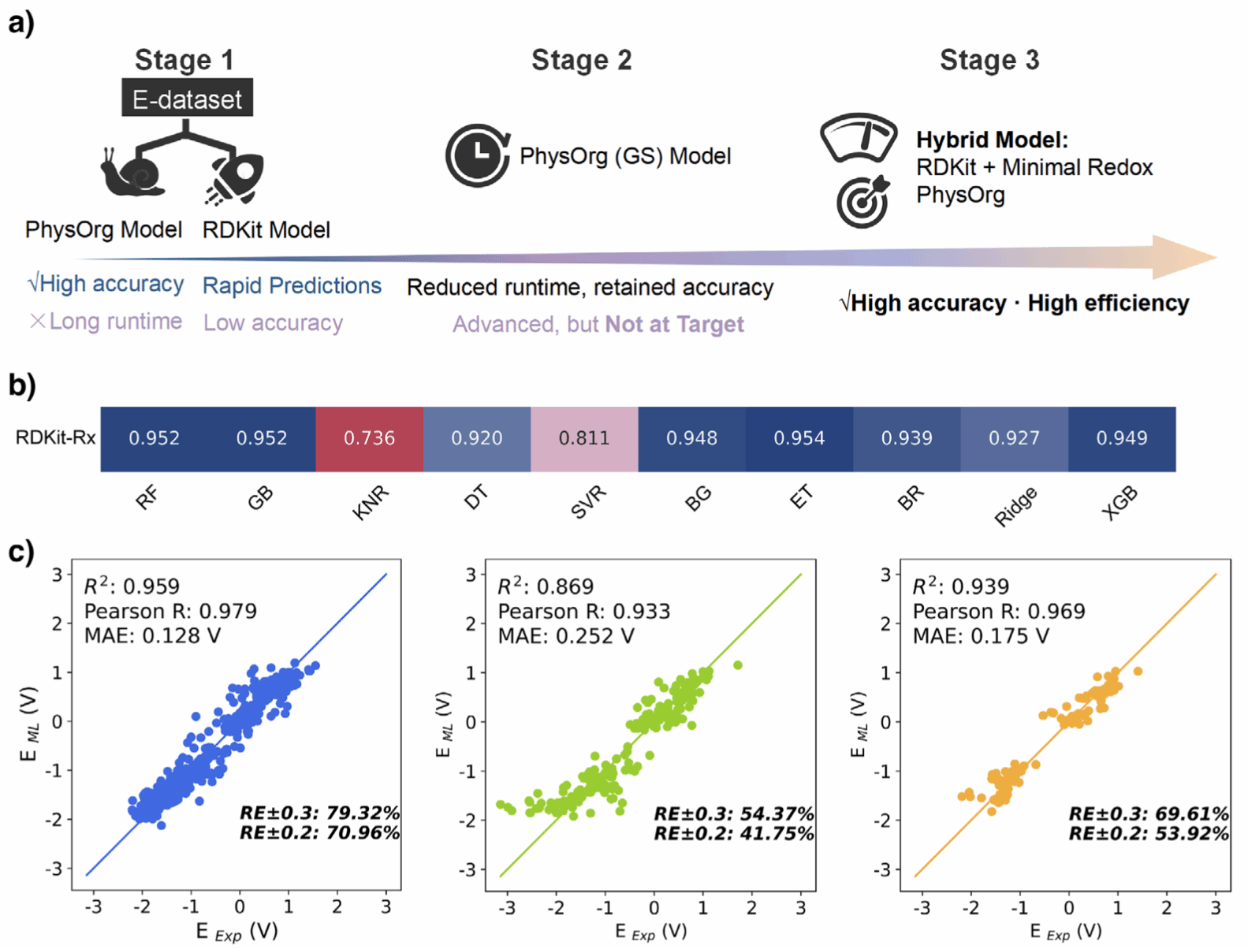
激发态模型(Model E)的优化与验证展示了激发态电位预测模型从构建到优化的全过程,展示了激发态模型在保持高精度的同时大幅降低计算成本,适用于高通量筛选。(a):三阶段优化流程——比较不同特征组合、替换高成本电子态特征、开发高效RDKit-Rx特征集。(b):不同算法与特征集性能对比,ET模型表现最佳。(c):最终Model E在交叉验证、外样本和外推数据上的预测结果,R⟡约0.87–0.96,MAE低至0.25 V。

为验证模型的普适性,团队进一步开展跨金属迁移学习研究:以铱(III)模型为基础,对少量锇(Os) 光催化剂数据进行残差迁移学习(Residual Transfer Learning);迁移模型(G-T、E-T)在仅需极少样本的情况下,预测精度(MAE≈0.2 V,R⟡≈0.9)已接近专属Os模型。这都表明该框架可在不同过渡金属体系间高效迁移与重用,显著降低新材料预测成本,展示了跨体系泛化能力。
该研究提出的机器学习–量子化学融合策略,不仅实现了对铱(III)光催化剂基态与激发态氧化还原电位的高精度、低成本预测,还揭示了电子结构对氧化还原性能的关键影响规律。研究成果为光催化剂的高通量筛选与理性设计提供了可解释、可迁移的计算框架,对推动光氧化还原催化、太阳燃料生产及绿色能源技术的发展具有重要意义。
该成果以“Machine Learning-Assisted Prediction of Ground- and Excited-State Redox Potentials in Iridium(III) Photocatalysts”为题,在国际知名期刊《Angewandte Chemie International Edition》上发表。东北师范大学化学学院博士研究生李雪涛为该文章的第一作者,通讯作者是化学学院教师关威教授与朱博副教授。
该研究获得了国家自然科学基金委员会的资助(项目编号:22173016、22203014),研究由东北师范大学化学学院功能材料化学研究所完成。
原文链接:https://onlinelibrary.wiley.com/doi/10.1002/anie.202517393
Copyright 2017 © 东北师范大学化学学院 All Rights Reserved 地址:吉林省长春市人民大街5268号 邮编:130024 电话:0431-85099667

